Exploring the genomics diversity gap
Nishant Panicker, B.S.
March 2026
(13 Minutes)
In 2016, a team of researchers at Harvard and the Broad Institute reanalyzed genetic data from patients who had been told they carried mutations for hypertrophic cardiomyopathy, a condition in which the heart muscle thickens and can cause sudden cardiac death (Manrai et al. 2016). What they found should have caused a stir in the scientific community – multiple variants classified as disease-causing in standard databases were, in fact, benign. They were common, harmless mutations called polymorphisms that have been observed frequently in populations of African ancestry. The patients who carried these variants had been misdiagnosed, some had undergone invasive procedures and others had lived for years under the presumed guise of a condition they never had. Where did this error come from?
The error did not originate in any single laboratory, but instead was built into the foundations of the field itself. The reference databases against which these variants were judged had been constructed predominantly from the genomes of individuals with European ancestry, and what looked rare and dangerous against that backdrop was simply misidentified. For over two decades, the scientific community has been building an increasingly detailed map of the human genome and its links to disease. But what if this map was drawn almost entirely from a single neighborhood? The diversity gap in genomics refers to the persistent underrepresentation of non-European populations in genome-wide association studies (GWAS), large-scale genetic studies that search for links between specific genes and diseases. It is not merely a demographic imbalance in research participation, but a structural issue that reframes the lens through which we study the human genome.
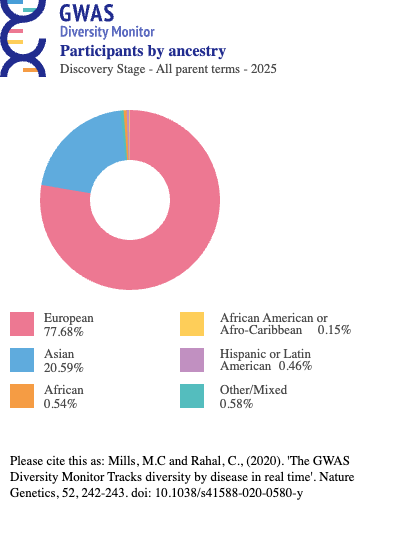
In 2009, Need and Goldstein conducted the first quantitative review of ancestral diversity in GWAS and found that 96% of participants were of European descent (Need and Goldstein 2009). By 2016, Popejoy and Fullerton’s work in Nature reported this figure had gone down to 81%, with individuals of Asian ancestry comprising 14% and those of African ancestry a mere 3% (Popejoy and Fullerton 2016). By 2021, the proportion with European ancestry climbed up to 86% (Mills and Rahal 2020). For a more recent depiction of the ancestry distribution in GWAS, see Figure 1, and if you are further interested visit https://gwasdiversitymonitor.com/. In terms of geographical concentration, 72% of participants in these studies were from just three countries – the United States, the United Kingdom and Iceland (Mills and Rahal 2019). In other words, the map of the human genome and its links to disease has been drawn almost entirely from a single neighborhood.
The origins of this imbalance
When genomics research began to accelerate in the late 20th century, the institutions with the funding, infrastructure and biobanking systems necessary for large-scale genetic studies were concentrated in Europe and North America. Early landmark projects like the Human Genome Project or the International HapMap Project drew from populations that were in close proximity to these centres. European-ancestry genomes thus formed the foundation of reference databases, and subsequent studies used those databases to carry out their analyses, producing results that reinforced the utility of European-ancestry data. This turned into a self-perpetuating cycle.
However, the structural origins of this imbalance are only a part of the reason it exists. There is a deeper explanation as to why the communities most underrepresented in genomic databases are often the ones most averse to participating in research studies, and it has nothing to do with scientific literacy or cultural attitudes toward medicine. Instead, it involves a legacy of unethical research practices that have fostered mistrust of the scientific establishment among marginalized communities.
When the map fails
Consider what happens when a physician orders a genetic test for a patient whose ancestry is not well represented in reference databases. The lab sequences the relevant gene, identifies a variant, and searches for it in the reference databases. If the variant appears frequently in well-studied populations and has been observed alongside disease, it is classified as pathogenic. If it appears frequently but has been unlinked to disease, it is classified as benign. However, if there isn’t enough data to sufficiently determine whether it is pathogenic or benign, it receives a third classification, a variant of unknown significance. With this result having no actionable information for a physician, the patient is left in a diagnostic limbo.
A large cohort study of 1.7 million people undergoing multigene panel testing found that variants of unknown significance were significantly more prevalent in Hispanic, African American, Asian and Pacific Islander populations compared to White patients (Chen et al. 2023). The underlying reason is straightforward – with far more extensive genomic information available for people of European ancestry, labs have a richer body of evidence to classify variants from these populations, while variants from underrepresented groups remain ambiguous.
Predicting risk for some, guessing for others
Polygenic risk scores represent an ambitious application of modern genomics. By aggregating the small effects of thousands of genetic variants, researchers aim to predict a person’s susceptibility to complex diseases like diabetes, cardiovascular disease and cancer. The strength of this approach sounds promising – a single score that could theoretically flag an individual’s disease risk years before symptoms show up, allowing for early intervention and targeted prevention.
But the algorithms that generate these risk scores were trained predominantly on genomic information from individuals of European ancestry, and their performance degrades dramatically across ancestral boundaries. Risk predictions in populations of African ancestry show reductions in accuracy of up to 78% compared to European populations, with substantially lower accuracy on average in Hispanic, South Asian, East Asian and African American populations (Martin et al. 2019; Duncan et al. 2019).
The clinical implication of this outcome is not trivial.
Most lung cancer polygenic risk score models, for instance, have been developed in European populations, where differences in gene variant frequencies and genetic architecture lead the scores to fail in identifying high-risk subpopulations in Chinese populations. Melanoma and prostate cancer models show similarly reduced predictive power in sub-Saharan African populations. If the promise of precision medicine is to reach everyone, the genomic foundation upon which it is built must include everyone.
Drugs don’t treat everyone equally
If the genome shapes disease risk, it also shapes how the body processes the drugs meant to treat those diseases. Pharmacogenomics, the study of how genetic variation affects drug response, has been disproportionately studied in European populations and the consequences of this imbalance are measured in adverse events and treatment failures. Is this beginning to sound familiar?
A compelling example is that of warfarin, one of the most widely prescribed anticoagulants in the world. African Americans and Latinos demonstrate greater variability in warfarin dose requirements and face an elevated risk of adverse events compared to individuals of European ancestry (Asiimwe and Pirmohamed 2022). The vast majority of literature on genotype-guided warfarin dosing was developed in European-ancestry populations, making the translation of these dosing guidelines to non-European patients unreliable.
Another example involves the enzyme CYP2C19, which plays a crucial role in metabolizing clopidogrel, a drug commonly prescribed to prevent blood clots after heart attacks. The frequency of CYP2C19 variants that impair drug metabolism differs markedly across populations – the CYP2C19*2 allele, associated with reduced drug efficacy, occurs at a frequency of approximately 32% in South Asian populations and 29% in East Asian populations, compared to 15% in European populations (Fricke-Galindo et al. 2016). This means that roughly twice as many South and East Asian patients carry a variant that makes this drug less effective – yet the dosing guidelines were derived primarily from European populations.
There is a pattern that emerges as we weave through these different consequences of the genomics diversity gap, and it is worth pausing to take a clear look. Variants of unknown significance leave patients without answers. Misclassified variants like the hypertrophic cardiomyopathy case described at the beginning of this article give patients the wrong answers. Inaccurate polygenic risk scores give them either false reassurance or false alarm. And pharmacogenomic blind spots can lead to inappropriate dosing of the right drug, or dosage of a drug their body cannot use. The progression follows the entire arc of genomic medicine, from diagnosis to prognosis to treatment, and at every stage, the patients most underserved are those whose ancestries the field does not represent.
The historical legacy of mistrust and medical malpractice
To provide one explanation as to why the diversity gap persists despite widespread acknowledgment of its consequences, it is important to recognize the complicated history and legacy of unethical medical research practices that have plagued marginalized communities.
In 1932, the United States Public Health Service enrolled nearly 600 African American men in Macon County, Alabama, into what would become one of the most notorious studies in the history of medicine, the Tuskegee syphilis study. The men – 399 with syphilis and a control group of 201 – were told they were receiving treatment for “bad blood”. They were not. They underwent painful procedures including spinal taps under the guise of therapy. When penicillin was established as an effective treatment for syphilis in 1945, the study’s investigators did not offer it to them. The study continued for another 27 years. It was not terminated because of an internal ethical review, but instead because a whistleblower went to the press in 1972. The disclosure is correlated with population level increases in medical mistrust and mortality among African American men, and the reverberations have been offered as one explanation for the persistent underrepresentation of Black Americans in research trials decades later.
Another such story is that of Henrietta Lacks, a 31 year old African American woman being treated for cervical cancer at Johns Hopkins Hospital. During her treatment, cells were taken from her cervix without her providing consent. The cells, which proved capable of dividing indefinitely in culture, became one of the most consequential tools in the history of biomedical research, the HeLa cell line. HeLa cells contributed to the development of the polio vaccine, cancer therapies, IVF technology and countless other scientific advances. While biotech companies sold the cells commercially for decades, Henrietta herself died in 1951 while her family lived in poverty, struggled to afford health insurance, and didn’t learn of the cell line’s existence until more than 20 years after her death. In 2013, researchers published the complete genetic sequence of HeLa cells without consulting the family, raising important questions about genetic privacy and consent. The Lacks family finally reached a settlement with Thermo Fisher Scientific in 2023 – seventy-two years after the cells were taken.
These are just two stories that happened to receive widespread coverage – what about the countless others whose stories were not picked up by the mainstream media? It is clear that these experiences are not simply relics of a distant era. They are part of the living memories of marginalized communities. The diversity gap is thus not only a problem of infrastructure and funding, but also one of the socio-historical context influencing trust in the very field of medicine itself.
Closing the diversity gap requires more than just recruitment targets for research studies. It requires transparency about how genetic data will be used and stored, and governance structures that give communities meaningful control over their own genetic information. The scale of this problem has certainly not gone unrecognized.
Working toward building a wider map
The Human Heredity and Health in Africa (H3Africa) consortium has developed a 2.3 million SNP array designed to capture the genetic diversity across the continent’s numerous ethnolinguistic groups, addressing a critical gap in the genomic tools available for African populations. The GenomeAsia 100K Project aims to sequence 100,000 whole genomes spanning multiple population groups across 64 countries in Asia. In the United States, the National Institute of Health’s All of Us research program has enrolled over 400,000 participants, with 46% belonging to minority racial or ethnic groups, and has shared the most extensive pool of African American, Hispanic and Latin American genomes to date. These are important advances in the field and are already producing results. When Egyptian patients with hypertrophic cardiomyopathy were reclassified using ancestry-matched controls, the diagnostic test results improved measurably. This single rectification, made possible by a more representative database, means that patients who would have been left in diagnostic uncertainty now receive clear answers. Imagine the scale of what a wider map could accomplish.
But the most promising sign may not be technological at all. It is the growing recognition, within the field itself, that the path forward requires a fundamentally different relationship between research institutions and the communities they study. The All of Us program was not designed merely to collect diverse genomes – it was designed with community advisory boards, transparent data governance and a commitment to returning results to participants.
The technology to sequence a genome is already reasonably priced and getting cheaper. The trust to hand one over must be earned, and earning it is harder work. But it is work that has begun. Each correction is quiet, incremental, and largely invisible to the patients it protects – a variant reclassified, a dosing algorithm recalibrated, a risk score that finally accounts for every individual. None of these will make headlines. But collectively, they are the difference between a field that promises precision for everyone and one that actually delivers it.
References

Leave a comment